

Novinky
V rámci optimalizace instrumentační metody na Synapt G2 TOF jsme v posledních dnech udělali několik vylepšení.
- Snížení energie aplikované na ionty při přechodu ze zdroje do vakua (nově standardně nižší Cone Voltage, Extraction Cone Voltage, teplota desolvačního plynu a napětí na sprejovací kapiláře), pro omezení fragmentace proteinů v plynné fázi
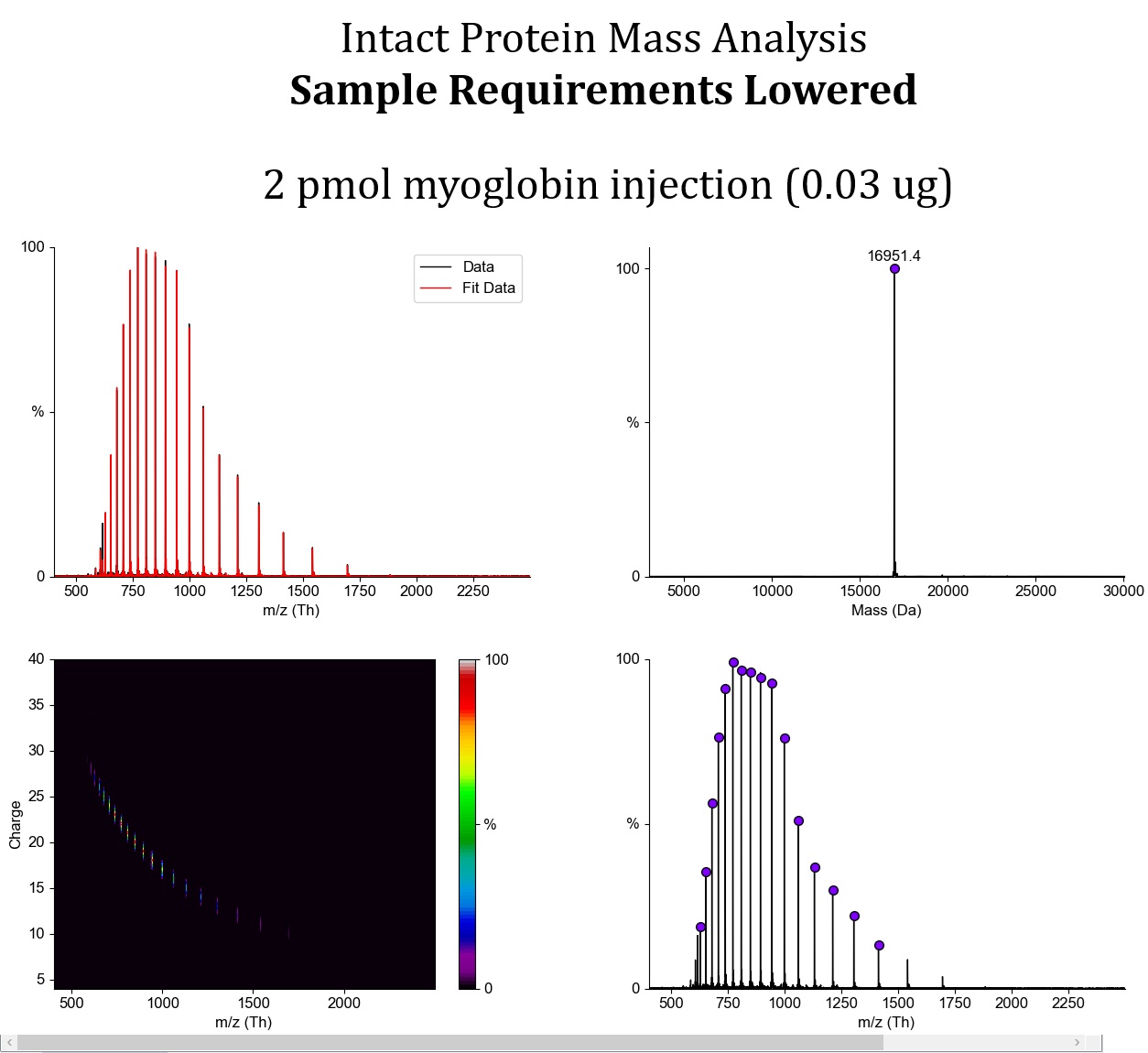
- Zjištění limitu detekce na modelovém proteinu, úprava našeho interního QC na 5x nižší množství proteinu, úprava doporučeného minimálního množství vzorku z μg/μl na molaritu (dává větší smysl v rámci MS), ve výsledku na 20 μl proteinu o koncentraci 1 μM (podle velikosti proteinu 30x-5x nižší než současné doporučení)
Za poslední rok a půl jsme provedli téměř 2000 měření intaktní hmoty proteinů. Vzhledem k dobře fungující rutině a obecné spokojenosti uživatelů jsme neměli potřebu výrazně optimalizovat nastřikované množství proteinů. Nicméně poslední dobou jsme se několikrát setkali s požadavkem na analýzu méně koncentrovaného vzorku, než který jsme doporučovali a zjistili jsme, že někdo kvůli tomu k analýze ani nepřistoupil a neoslovil nás.
Prozkoumali jsme proto důkladně historicky používané množství proteinů v intaktní analýze, prozkoumali skutečnou citlivost našeho LC-MS systému a upravili náš QC standard. Výsledek je, že doporučované množství proteinu pro intaktní analýzu je nyní výrazně nižší. Budeme snižovat nastřikované množství proteinů na cca 5 pmol, což může znamenat až 30x nižší koncentraci, podle velikosti analyzovaných proteinů. Do teď jsme také uvažovali a doporučovali množství vzorků v μg/μl, což není pro MS praktické. Náš signál je závislý na fyzickém množství molekul v roztoku a tomu odpovídá jedině molarita roztoku. Pro větší proteiny bude tedy identický požadavek na 1 μM vzorek v μg/μl vždy vyšší, než pro malý protein.
Také jsme si všimli, že některé proteiny našich uživatelů byly výrazně náchylnější na fragmentaci v plynné fázi, než jiné. Plošně proto snižujeme energii iontů při přechodu ze zdroje do hmotnostního spektrometru, abychom se vyhnuli jejich fragmentaci. Na modelovém myoglobinu jsme v zásadě kompletně eliminovali jakékoli fragmentace a hmotnostní spektrum nyní obsahuje jen signál intaktního proteinu. V naší dosavadní metodě docházelo k mírné fragmentaci, která se projevovala dodatečnými signály o nízké hmotě v oblasti nízkých m/z. Tato fragmentace pak byla u některých specifických uživatelských vzorků relativně výrazná.