

News
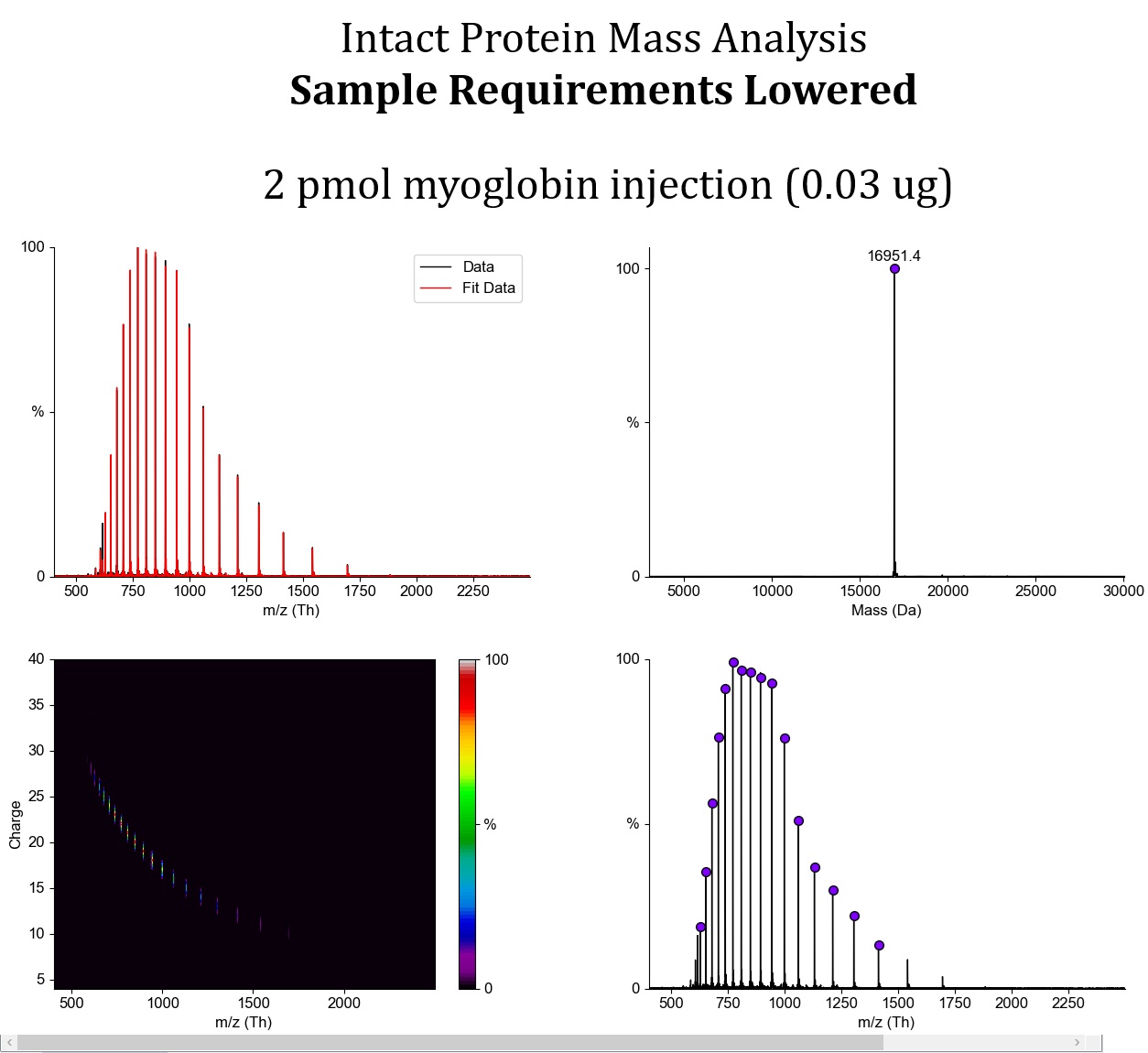
As part of optimizing the instrument method on the Synapt G2 TOF, we have made several improvements over the past few days.
- We reduced the energy applied to ions during transfer from the source into the vacuum region. This now means lower default settings for Cone Voltage, Extraction Cone Voltage, desolvation gas temperature, and spray capillary voltage, in order to reduce gas-phase fragmentation of proteins.
- We determined the limit of detection using a model protein, adjusted our internal QC to work with a 5-fold lower amount of protein, and changed our recommended minimum sample amount from µg/µL to molarity, which is more meaningful for MS. As a result, the new recommendation is 20 µL of protein at 1 µM concentration, which depending on protein size is roughly 5- to 30-fold lower than our previous recommendation.
Over the past year and a half, we have performed nearly 2,000 intact protein mass measurements. Because the workflow was functioning well and users were generally satisfied, we did not previously feel a strong need to further optimize the amount of protein injected. Recently, however, we encountered several requests to analyze samples that were less concentrated than what we had been recommending, and we also found out that in at least one case someone did not proceed with the analysis at all and did not contact us because of this.
We therefore carried out a thorough review of the protein amounts historically used for intact-mass analysis, evaluated the actual sensitivity of our LC-MS system, and updated our QC standard. The outcome is that the recommended amount of protein for intact analysis is now significantly lower. We will now reduce the injected amount to approximately 5 pmol, which may correspond to up to a 30-fold lower sample concentration, depending on the size of the protein being analyzed.
Until now, we had also been thinking about and recommending sample amounts in µg/µL, which is not very practical for MS. Our signal depends on the actual number of molecules present in solution, and that is properly reflected only by the molar concentration. For larger proteins, the same requirement of a 1 µM sample will therefore always correspond to a higher value in µg/µL than it would for a small protein.
We also noticed that some user proteins were much more prone to gas-phase fragmentation than others. We are therefore lowering the ion energy across the board during transfer from the source into the mass spectrometer in order to avoid this fragmentation.Using model myoglobin, we were able to essentially eliminate fragmentation completely, and the mass spectrum now contains only the signal of the intact protein. With our previous method, there was slight fragmentation, visible as additional low-mass signals in the low m/z region. In some specific user samples, this fragmentation was relatively pronounced.