

HDX-MS (Hydrogen/Deuterium Exchange Mass Spectrometry)
Method Overview
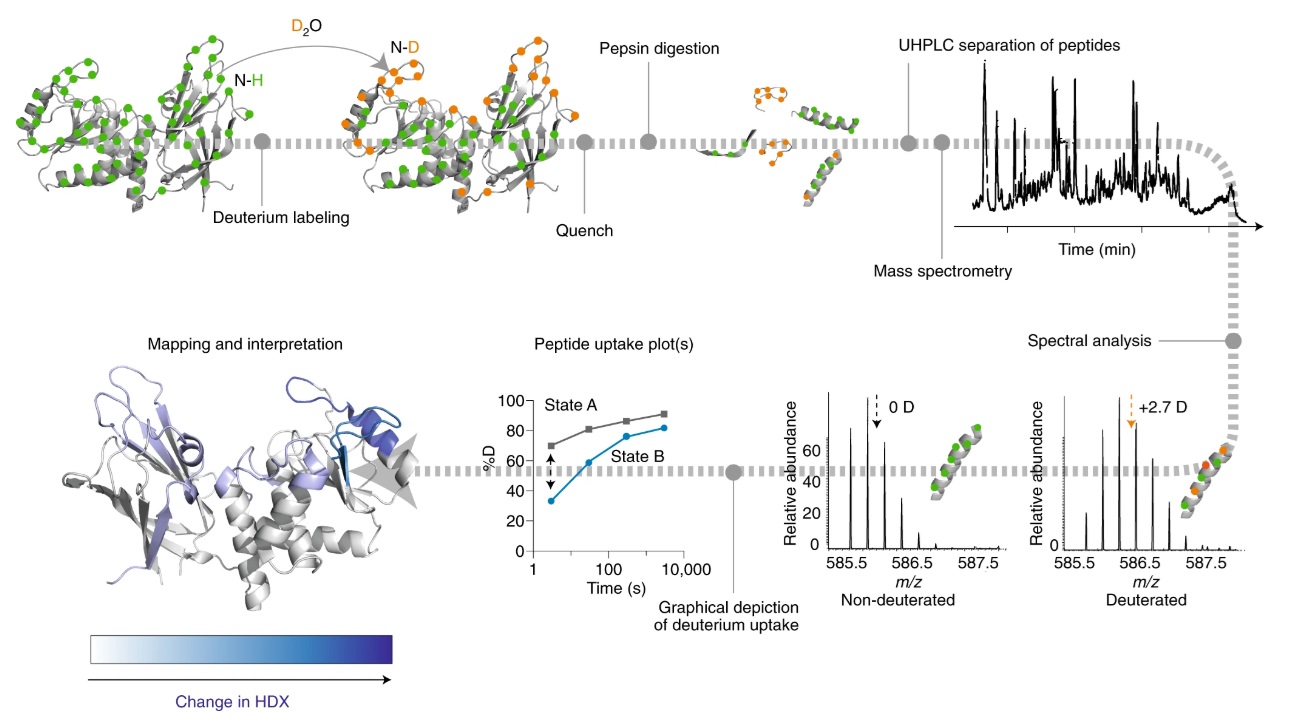
This method is based on measuring mass differences induced by exchange of peptide backbone hydrogen in proteins for deuterium from solvent. Because solvent accessibility and local hydrogen bonding influences the rate of exchange and this exchange happens on a timescale of seconds to hours, differential experiments can be performed to localize areas of protein-protein or protein-ligand interaction, or virtually any other effect that results in a change in local hydrogen bonding or local solvent accessibility.
Importantly, proteins can be analyzed using HDX-MS in their typical buffers with salts or other small molecules, so the resulting effects can be observed in a more native state compared to NMR or X-ray crystallography, which can provide similar information.
This method is a reliable way to characterize small molecule binding sites, antibody epitopes, strong protein-protein interaction interfaces, or even protein-nucleic acid binding interfaces. Structural changes on proteins due to temperature or pH changes can also be elucidated using HDX-MS.
Contact person: František Filandr
Requirements:
- Hypothesis of effect to observe and detailed sample information
- At least 100 ug of protein for each condition you would like to compare (with / without ligand)
- Protein in a buffer free of detergents (unless necessary for protein structure), ideally HEPES or TRIS
- Protocol of upstream sample handling, so we know what contaminants to expect in the samples
- Protein sequences
- Protein structural model, if available (can be AlphaFold)
Results:
- Map of areas of the protein, that show changes in deuterium uptake in a specific condition
- Model (if provided) with visualized deuterium uptake differences
- Help with interpretation of the results - elucidation of protein-protein interaction interface, protein dynamics or ligand binding site
Expected turnaround time:
- 2-3 weeks
If you are interested in HDX-MS, contact us.